Case of the Week #603
(1) Private sector; (2) King Husein Medical center; (3) Hashemite University
35-year-old G7P6 woman involved in a consanguineous marriage with a maternal relative was referred at 19 weeks, 2 days for detailed scan. Her obstetric history was significant for two miscarriages, and two infants (both a male and female) delivered with congenital anomalies who died soon after birth. The couple had a healthy girl and boy. Our prenatal imaging showed the following findings:
View the Answer Hide the Answer
Answer
We present a case of Meckel Gruber syndrome.
Our imaging, which was limited because of maternal body habitus (high BMI) and oligohydramnios, showed:
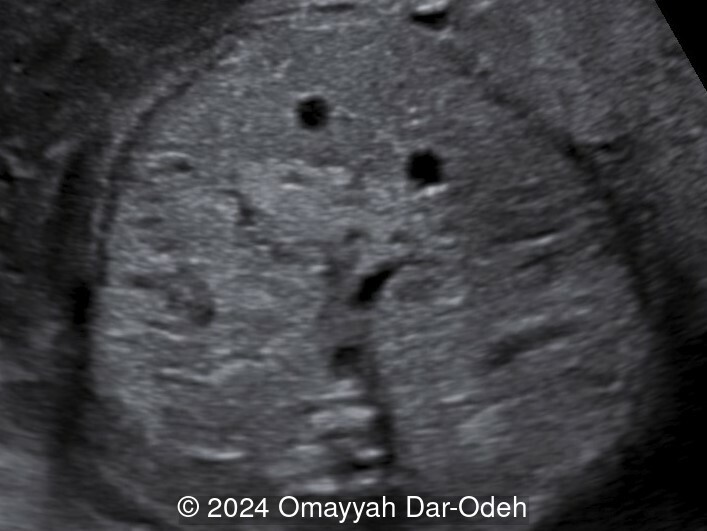
- Image 1: Transverse section in abdomen showing bilateral enlarged cystic kidneys
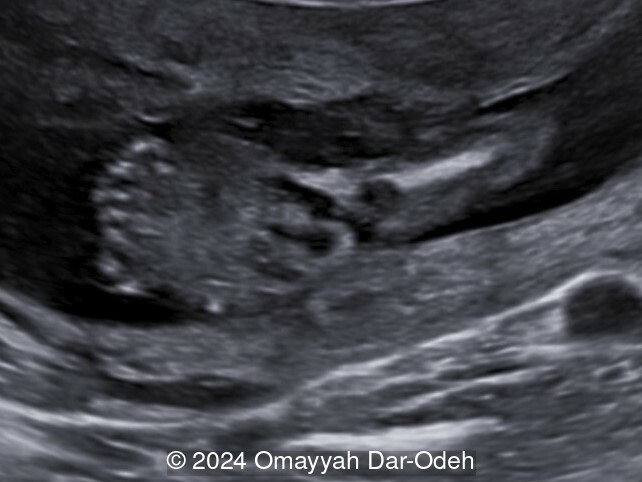
- Image 2: Postaxial polydactyly in foot
- Video 1: Postaxial polydactyly in hand
- Video 2: Abnormal skull shape and occipital encephalocele
- Video 3: Enlarged cystic kidneys
- Video 4: Club foot
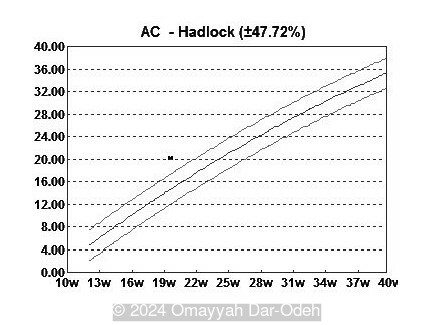
- Image 3: Large abdominal circumference
We suspected Meckel Gruber syndrome and advised the patient to consider amniocentesis for genetic study. Amniocentesis was delayed due to poor patient compliance and was technically difficult due to anhydramnios. Therefore, chorionic villous sampling was done.
Whole exome sequencing demonstrated a homozygous c.513C>G / p. Tyr171Ter class 2 variant in the B9D2 gene (B9 Domain Containing 2), which is a protein coding gene expected to cause B9D2 Related Disorder Phenotype (OMIM: 611951). Conditions associated with B9D2 include Meckel Syndrome, Type 10 and Joubert Syndrome 1. The patient was counselled regarding the lethal outcome. Genetic screening and clinical evaluation in family members were recommended in order to determine the pathogenic effect of this change.
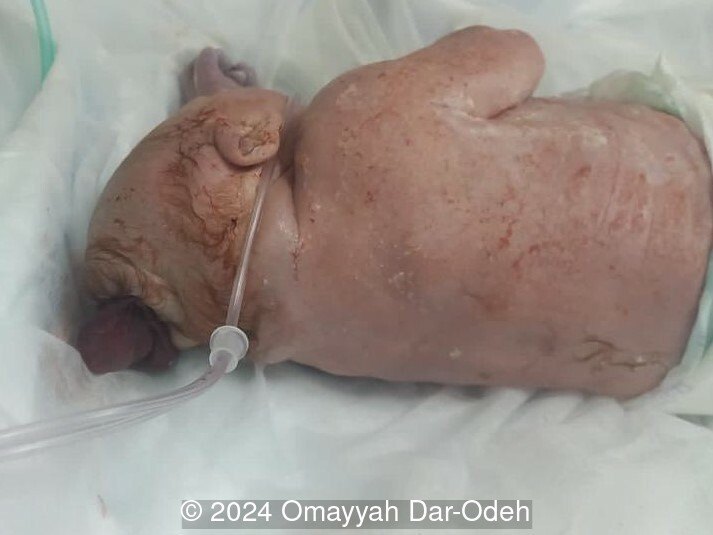
The couple refused termination and were followed at an outside hospital. The patient continued the pregnancy until 38 weeks and underwent lower uterine segment cesarean section due to three prior cesarean sections. A live male fetus weighing 2400 grams was delivered with multiple anomalies including microcephaly, large occipital encephalocele, distended abdomen, postaxial polydactyly in hands and feet. Soon after birth, the newborn had difficulty breathing with cyanosis and died 3 hours later. Autopsy was denied by the parents.
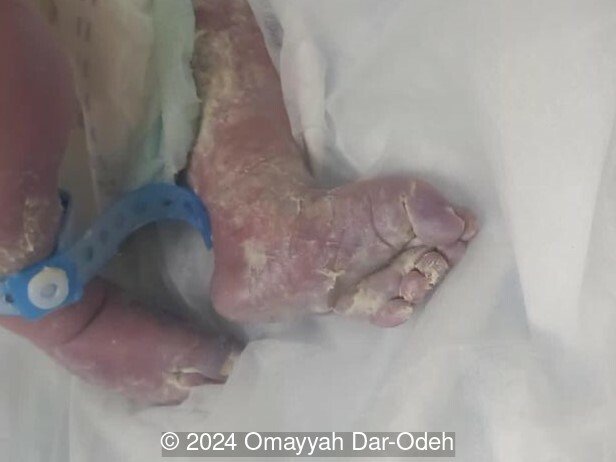
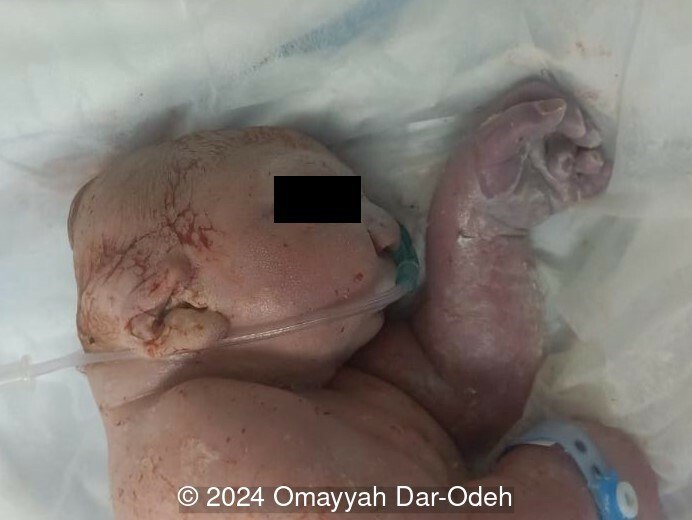
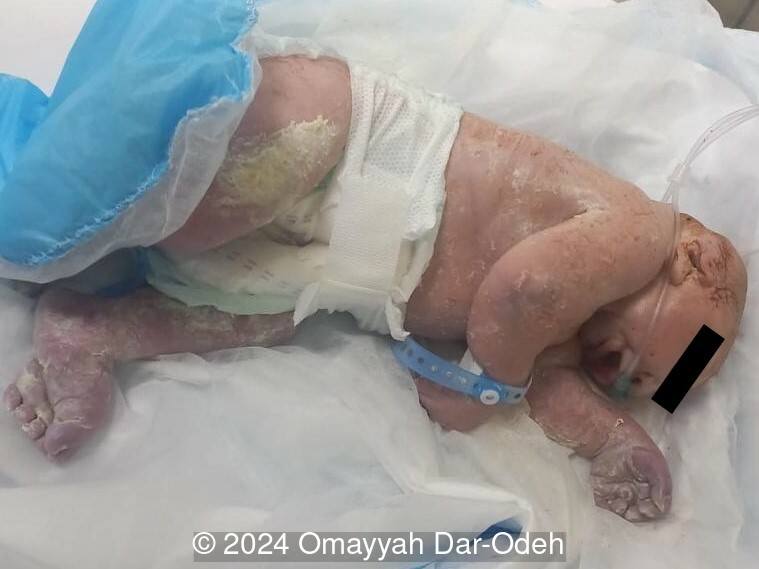
Discussion
Meckel–Gruber syndrome (MKS) is a rare, lethal and autosomal recessive disease with a 25% recurrence rate [1,2]. It is caused by mutations in genes encoding proteins that are structural or functional components of the primary cilium. Conditions that are caused by mutations in ciliary genes are collectively termed the ciliopathies, and Meckel–Gruber syndrome represents the most severe condition in this group of disorders.
It was originally described by Johann Friedrich Meckel in 1822 [4]. Gruber later published reports of patients with the same constellation of anomalies in 1934 and gave it the name dysencephalia splanchnocystica. Worldwide, its incidence is 1 per 13,250–140,000 livebirths [5]. Higher incidences are observed in endogamous populations, such as Gujarati Indians, Tatars, Hutterites, and in Finland where 1 in every 9,000 live births is affected [3]. An increased incidence has been reported in regions where consanguineous marriages are common (with 1 affected birth per 1,300).
Meckel–Gruber syndrome is characterized by the triad of cystic renal disease, a central nervous system malformation, and polydactyly [6]. The most common findings include renal cystic dysplasia (95-100%), occipital encephalocele (60-80%), and postaxial polydactyly (55-70%) [7]. At least two of the major antenatal ultrasound findings should be present for diagnosis of this syndrome. In approximately 57% of the cases, the three findings can be seen together. In addition to these classical findings, central nervous system findings can also include Dandy-Walker malformation, Arnold Chiari malformation, microcephaly, and hydrocephalus. Other anomalies include eye defects, cleft palate/lip, hepatic duct malformation characterized by proliferation and fibrosis in the portal areas of the liver and bile ducts, pancreatic cysts and fibrosis, congenital heart disease, adrenal hypoplasia, and genitourinary malformations such as male genital organ hypoplasia, male pseudohermaphroditism, cryptorchidism, ureter agenesis, hypoplasia or duplication, and bladder agenesis and hypoplasia. Characteristic images on ultrasound are dependent on gestational age. While the classical triad can be better identified by ultrasound before the 14th week of pregnancy, it becomes difficult to diagnose with advanced oligohydramnios as gestation progresses.
Meckel–Gruber syndrome has extreme genetic heterogeneity and displays allelism with other ciliopathies such as Joubert syndrome, COACH syndrome (cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, ocular coloboma, and hepatic fibrosis), Oro-Facio-digital syndrome, nephronophthisis (NPHP), and Bardet–Biedl Syndrome [3]. To date, mutations in 14 genes are identified as causative for Meckel–Gruber syndrome. Genetic mutations on chromosome 17q21-24 are identified in most of the cases. Individual families with either Meckel–Gruber syndrome or MKS-like phenotypes have been found to have unique mutations [3]. In total, mutations in these genes appear to explain only 50–60% of Meckel–Gruber syndrome cases. Many of these genes are also responsible for Joubert syndrome, leading to the concept that Meckel–Gruber syndrome is the extreme lethal phenotype of Joubert syndrome [7]. Because of the genetic heterogeneity, whole genome sequencing can be helpful to identify the pathogenic mutation.
In our case, a mutation in B9D2 gene was identified. Domain-containing proteins Mks1, B9D1, and B9D2 interact physically and mutations may prevent B9D2 from interacting with Mks1, thus inhibiting B9D2 function. B9D1 is required for normal Hedgehog (Hh) signaling, ciliogenesis, and ciliary protein localization. B9D1 and B9D2 are essential components of a B9 protein complex and, when disrupted can result in Meckel -Gruber syndrome [8].
In cases with multiple anomalies (CNS, cardiac, renal and extremity), Trisomy 13 and Smith Lemli-Opitz syndrome should be considered in the differential diagnosis [1]. The prognosis in Meckel-Gruber syndrome is poor. It is often lethal due to dysplastic kidneys resulting in severe oligohydramnios and pulmonary hypoplasia [7].
References
[1] Erol SA, Kırbaş A. Prenatal Sonographic Diagnosis of Meckel-Gruber Syndrome: A Case Report. Med J West Black Sea. 2021;5(1):110-113.
[2] Raj M, Dhanuka S, Agarwal P, et al. Meckel Gruber syndrome – a case report. Surg Exp Pathol. 2020 April 15:3:11.
[3] Hartill V, Szymanska K, Sharif SM, et al. Meckel–Gruber Syndrome: An Update on Diagnosis, Clinical Management, and Research Advances. Front Pediatr. 2017; 5: 244.
[4] No Author. Johann Friedrich Meckel, the Younger (1781-1833). JAMA. 1970 Oct 5;214(1):138-9.
[5] Aslan K, Aslan EK, Orhan A, et al. Meckel Gruber syndrome, A case report. Organogenesis. 2015 Jun;11(2):87-92.
[6] Gazioğlu N, Vural M, Seçkin MS, et al. Meckel-Gruber syndrome. Childs Nerv Syst. 1998 Mar;14(3):142-5.
[7] Roy J, Pal M. Meckel Gruber Syndrome. J Clin Diagn Res. 2013 Sept;7(9):2102-2103.
[8] Kheir AEM, Imam A, Omer IM, et al. Meckel-Gruber syndrome: A rare and lethal anomaly. Sudan J Paediatr. 2012; 12(1): 93–96.
Discussion Board
We appreciate your patience as we review all submitted answers. Check back soon to see if you were correct!